In Von Hippel-Lindau Syndrome, abbreviated VHL, an individual has an abnormal genetic message coded by an identified gene located on the short arm of chromosome 3 caused by a germline mutation. This allows the development of abnormal blood vessel growth forming tumors, which can be single or multiple, and occur most commonly in the brain, spinal cord, eyes, kidneys, adrenal glands and inner ear. Cysts, which are fluid-filled sacs, can also occur in the kidney, central nervous system, liver, pancreas, adrenal and reproductive systems. VHL is highly associated with the development of renal cell carcinoma caused by the degeneration of renal cysts.
Everyone has the VHL gene. The improper function of that gene allows the growth of blood vessels. Normal vessels grow like trees; VHL vessels grow in knots called angiomas or hemangioblastomas. These angiomas in time damage neighboring tissue usually by rapid growth, less so by bleeding, and depending on their location, cause patients to present with symptoms such as blindness, neurologic problems of balance, weakness and headaches, alterations of blood pressure and fainting or in late stages outright signs of disseminated cancer. Early detection and careful monitoring employing imaging modalities of CT, MRI and ultrasound are necessary to insure proper care of the patient.
MR surveillance in a patient with VHL disease shows hemangioblastomas (arrow) in (A) Brain and (B) Spine.
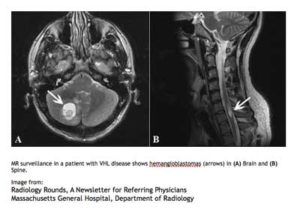
Image from:
Radiology Rounds, A Newsletter for Referring PhysiciansMassachusetts General Hospital, Department of Radiology
HISTORY
Dr. Eugen von Hippel described the angiomas in the eye in 1904. His name is usually associated with VHL in the retina, though in his original case reports, he found patients with tumors in other locations.
Dr. Arvid Lindau described the angiomas of the cerebellum and spine in 1926. His name is usually associated with occurrence of VHL in the central nervous system.
In 1994, the VHL gene was identified as well as the structure of the VHL protein (tumor suppressor protein) and HIF (hypoxic inducible factor protein). Within the next few years the genetic correlation with kidney cancer and cerebellar hemangioblastomas in VHL patients with malfunction of this gene was recognized.
INHERITANCE
Most commonly, VHL is a genetically transmitted condition caused by a germline mutation in a dominant gene and passed on from one parent to progeny (see diagram). In 20% of cases, the disease is spontaneous and neither parent nor any relative possess the gene abnormality or disease manifestation. Therefore, everyone in the population has some risk for this illness which has an incidence of 1:32,000. The risk is much higher in those with a family history, a 50% chance in offspring in a family where one parent has the dominant gene.
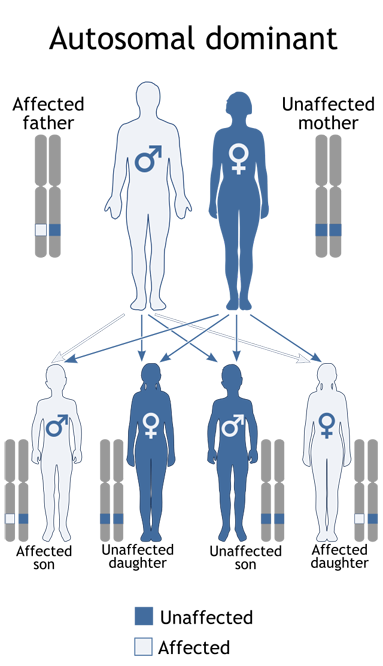
Inheritance of VHL
A child receives one gene in each pair from each parent. If one parent has a Dominant gene (D), each child has a 50-50 chance of inheriting the condition. Dominant genes dominate their normal counterparts (n). A dominant gene can be inherited by either a male or a female child.
DETECTION AND TREATMENT
With a family history for VHL, screening in progeny should begin by the age of six or earlier with ophthalmoscopy in infancy in at risk groups. Screening should be performed under the guidance of a treating physician and/or genetic counselor experienced in managing VHL. DNA testing is the definitive diagnostic test. Even without symptoms in their VHL parents, children are at risk and should be genetically screened. Once a genetic diagnosis has been made, a comprehensive serial screening protocol and routine scheduled follow-ups are essential for proper care. Presenting initial symptoms have manifested as early as the age of 1 and as late as the age of 84. There is a 90% penetrance by the age of 65 and morbidity is chiefly due to complications from renal cancer and CNS hemangioblastomas.
Symptoms of persistent headaches, dizziness, weakness, hearing loss, fluctuation in blood pressure or fainting or a finding of visual loss with an angioma should prompt the immediate appropriate evaluation and imaging in VHL patients.
Prior to comprehensive screening protocols, DNA diagnosis, and advances in treatment, median survival in VHL patients was less than 50 years of age. It has improved over the last 15 years, and with the promise of advances in biogenetics for targeted drug therapy, better biomechanical and radiologic treatments, and development of screening blood tests to measure disease activity, there is hope for additional benefit to extend and improve the quality of life for VHL patients. This can only be accomplished with continual financial support for research efforts in these areas.